Yet, there are some studies linking Zinc oxide to Alzheimers and I am not sure how much Zinc Gluconium (the key ingredient in Cold Eeze) translates into Zinc oxide flowing through my brain. Am I putting myself at risk by taking excessive amounts of zince 4-5 times per year?
Maybe.

Each Cold Eeze has 13.3mg of Zinc Gluconium. The upper limit recommended of Zinc is 40mg. The Cold Eeze box says use every 2-4 hrs until symptoms subside. If we assume I take it every 2 hrs, which I have done in the past, even sleeping with one in my mouth, which I definitely do NOT recommend (but I was desperate to not get sick), then my total for a 24hr period is 159.6mg. This is excessive.
Here are some recent studies looking into the role of Zinc in Alzheimers.
For now the key is to avoid going over 40mg per day. But will Cold Eeze truly work for me in this way? That is only 3 Cold Eeze a day. I will find out, but I am suspicious I need more Zinc lozenges to fight a cold. I still follow the bone broth/chicken soup intake I recommended in a previous post.

https://drcremers.com/2017/05/best-ways-to-fight-cold-and-from.html
And hot showers, massaging, etc. But will that be enough to fight a cold without excessive Cold Eeze?
To be continued.
SLC
Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease
 1,2
1,2Introduction
Results
Zn2+ promotes Aβ40 and Aβ42 oligomer formation

Zn2+ rapidly changes secondary structures of Aβ40 and Aβ42 and retains Aβ in a less β-conformation
ZnAβ40 and ZnAβ42 form oligomers that are less heterogeneous and possess higher hydrophobic-exposed surfaces than ADDLs

Low immunoreactivity of ZnAβ oligomers reveal distinct oligomeric structure
ZnAβ40 oligomers are structurally more disordered than Aβ40 fibrils

Removal of Zn2+ restores amyloid fibril formation with a faster kinetics and ZnAβ is unable to serve as fibrillization seeds


ZnAβ oligomers possess higher cytotoxicity than ADDLs

ZnAβ oligomers inhibit hippocampal LTP and increase microglial activation in wild-type mice


Discussion
Materials and Methods
Aβ synthesis and preparation
ThT Assay
Dot blotting
Direct blue staining
Far-UV CD spectroscopy
TEM
Bis-ANS
AUC
Solid-state NMR
Aβ seeding assay
Cytotoxicity assay
Calcium influx assay
Animal
In vitro electrophysiology
Acute Aβ-injected mice models
Immunohistochemistry
Data Availability
Acknowledgements
Author Contributions
Footnotes
References
The study below is also interesting, but it does not mention the biggest concern of all: the role of Zinc excess in Alzheimers.
——–
Plasma metals as potential biomarkers in dementia: a case–control study in patients with sporadic Alzheimer’s disease
Abstract
Keywords
Alzheimer’s disease Dementia Neurodegeneration Human plasma Metal biomarker Plasma-zinc levels
Abbreviations
Introduction
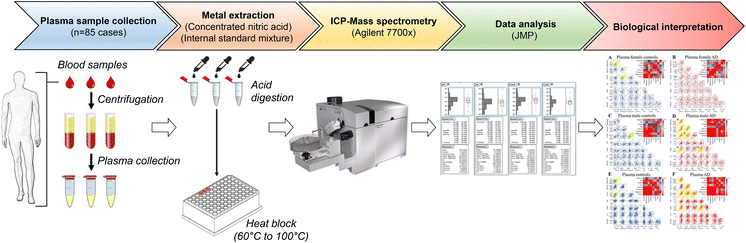
Materials and methods
Ethics
Patient selection
Sample digestion
Metal measurements
Statistical methods
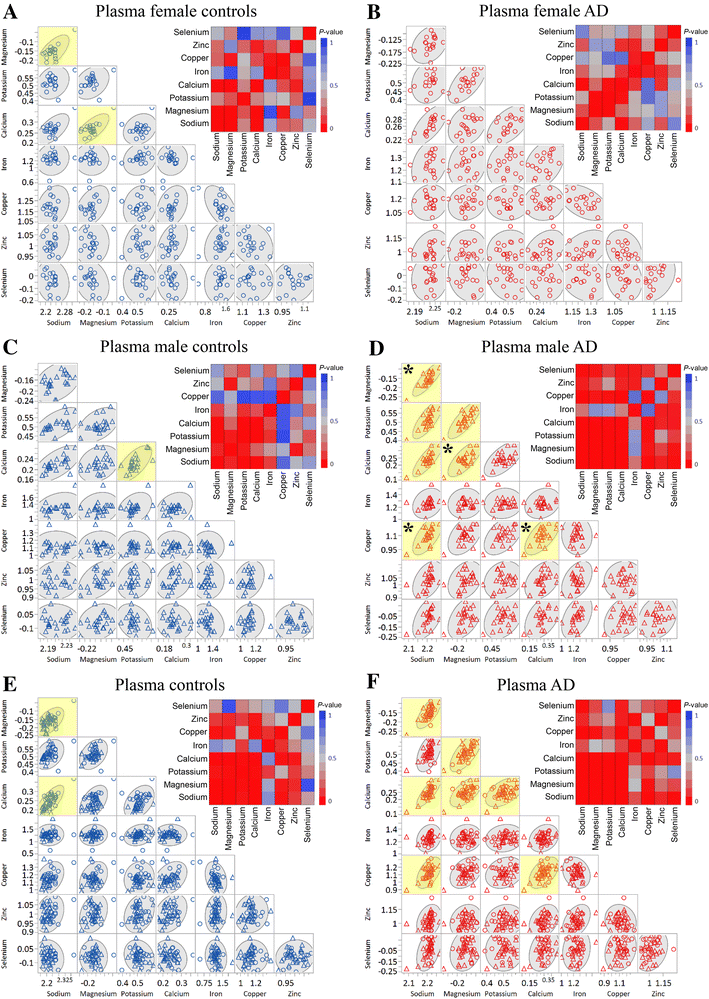
Results and discussion
Notes
Acknowledgements
Compliance with ethical standards
Conflict of interest
References
-
Adlard PA, Bush AI (2006) Metals and Alzheimer’s disease. J Alzheimer’s Dis 10:145–163CrossRefGoogle Scholar
-
Akatsu H et al (2012) Transition metal abnormalities in progressive dementias. Biometals 25:337–350. https://doi-org.ezp.welch.jhmi.edu/10.1007/s10534-011-9504-8CrossRefPubMedGoogle Scholar
-
Ayton S, Lei P, Bush AI (2013) Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med 62:76–89. https://doi-org.ezp.welch.jhmi.edu/10.1016/j.freeradbiomed.2012.10.558CrossRefPubMedGoogle Scholar
-
Bush AI (2013) The metal theory of Alzheimer’s disease. J Alzheimer’s Dis 33:S277–281. https://doi-org.ezp.welch.jhmi.edu/10.3233/jad-2012-129011Google Scholar
-
Church SJ et al (2015) Deficient copper concentrations in dried-defatted hepatic tissue from ob/ob mice: a potential model for study of defective copper regulation in metabolic liver disease. Biochem Biophys Res Commun 460:549–554. https://doi-org.ezp.welch.jhmi.edu/10.1016/j.bbrc.2015.03.067CrossRefPubMedPubMedCentralGoogle Scholar
-
Citron M (2010) Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov 9:387–398. https://doi-org.ezp.welch.jhmi.edu/10.1038/nrd2896CrossRefPubMedGoogle Scholar
-
Cooper GJS et al (2005) Demonstration of a hyperglycemia-driven pathogenic abnormality of copper homeostasis in diabetes and its reversibility by selective chelation. Diabetes 54:1468–1476. https://doi-org.ezp.welch.jhmi.edu/10.2337/diabetes.54.5.1468CrossRefPubMedGoogle Scholar
-
Craddock TJ, Tuszynski JA, Chopra D, Casey N, Goldstein LE, Hameroff SR, Tanzi RE (2012) The zinc dyshomeostasis hypothesis of Alzheimer’s disease. PLoS ONE 7:e33552. https://doi-org.ezp.welch.jhmi.edu/10.1371/journal.pone.0033552CrossRefPubMedPubMedCentralGoogle Scholar
-
Ferri CP et al (2005) Global prevalence of dementia: a Delphi consensus study. Lancet 366:2112–2117CrossRefPubMedPubMedCentralGoogle Scholar
-
Fiandaca MS, Mapstone ME, Cheema AK, Federoff HJ (2014) The critical need for defining preclinical biomarkers in Alzheimer’s disease. Alzheimer’s Dement 10:S196–212. https://doi-org.ezp.welch.jhmi.edu/10.1016/j.jalz.2014.04.015CrossRefGoogle Scholar
-
Gerhardsson L, Lundh T, Minthon L, Londos E (2008) Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord 25:508–515. https://doi-org.ezp.welch.jhmi.edu/10.1159/000129365CrossRefPubMedGoogle Scholar
-
Huang X et al (1999) The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38:7609–7616. https://doi-org.ezp.welch.jhmi.edu/10.1021/bi990438fCrossRefPubMedGoogle Scholar
-
Huang X, Moir RD, Tanzi RE, Bush AI, Rogers JT (2004) Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann NY Acad Sci 1012:153–163CrossRefPubMedGoogle Scholar
-
Huang CW, Wang SJ, Wu SJ, Yang CC, Huang MW, Lin CH, Cheng IH (2013) Potential blood biomarker for disease severity in the Taiwanese population with Alzheimer’s disease. Am J Alzheimer’s Dis Other Dement 28:75–83. https://doi-org.ezp.welch.jhmi.edu/10.1177/1533317512467674CrossRefGoogle Scholar
-
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53:474–481. https://doi-org.ezp.welch.jhmi.edu/10.2337/diabetes.53.2.474CrossRefPubMedGoogle Scholar
-
Kaddurah-Daouk R et al. (2013) Alterations in metabolic pathways and networks in Alzheimer/’s disease, Transl Psychiatry 3:e244. http://www.nature.com.ezp.welch.jhmi.edu/tp/journal/v3/n4/suppinfo/tp201318s1.html
-
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158:47–52CrossRefPubMedGoogle Scholar
-
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology 34:939–944CrossRefPubMedGoogle Scholar
-
Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J (2006) Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J Struct Biol 155:30–37. https://doi-org.ezp.welch.jhmi.edu/10.1016/j.jsb.2005.09.004CrossRefPubMedGoogle Scholar
-
Nahan KS, Walsh KB, Adeoye O, Landero-Figueroa JA (2017) The metal and metalloprotein profile of human plasma as biomarkers for stroke diagnosis. J Trace Elem Med Biol 42:81–91. https://doi-org.ezp.welch.jhmi.edu/10.1016/j.jtemb.2017.04.004CrossRefPubMedGoogle Scholar
-
Nittis T, Gitlin JD (2002) The copper-iron connection: hereditary aceruloplasminemia. Semin Hematol 39:282–289CrossRefPubMedGoogle Scholar
-
Prince M, Guerchet M, Prina M (2013) Policy brief for heads of government: the global impact of dementia 2013–2050. Alzheimer’s Dis Int 2013:1–8Google Scholar
-
Ramos P, Santos A, Pinto NR, Mendes R, Magalhaes T, Almeida A (2014) Anatomical region differences and age-related changes in copper, zinc, and manganese levels in the human brain. Biol Trace Elem Res 161:190–201. https://doi-org.ezp.welch.jhmi.edu/10.1007/s12011-014-0093-6CrossRefPubMedGoogle Scholar
-
Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA (2012) The role of metallobiology and amyloid-beta peptides in Alzheimer’s disease. J Neurochem 120:149–166. https://doi-org.ezp.welch.jhmi.edu/10.1111/j.1471-4159.2011.07500.xCrossRefPubMedGoogle Scholar
-
Sastre M, Ritchie CW, Hajji N (2015) Metal ions in alzheimer’s disease brain. JSM Alzheimer’s Dis Relat Dement 2:1014Google Scholar
-
Schrag M et al (2011) Effect of cerebral amyloid angiopathy on brain iron, copper, and zinc in Alzheimer’s disease. J Alzheimer’s Dis 24:137–149. https://doi-org.ezp.welch.jhmi.edu/10.3233/jad-2010-101503Google Scholar
-
Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochem Biophys Acta 1768:1976–1990. https://doi-org.ezp.welch.jhmi.edu/10.1016/j.bbamem.2007.02.002CrossRefPubMedGoogle Scholar
-
Squitti R (2012) Metals in alzheimer’s disease: a systemic perspective. Front Biosci 17(1):451CrossRefGoogle Scholar
-
Vardy ER, Rice PJ, Bowie PC, Holmes JD, Grant PJ, Hooper NM (2007) Increased circulating insulin-like growth factor-1 in late-onset Alzheimer’s disease. J Alzheimers Dis 12:285–290
https://www.health.harvard.edu/blog/zinc-for-the-common-cold-not-for-me-201102171498
Zinc for the common cold? Not for me

Other Articles waiting to access: