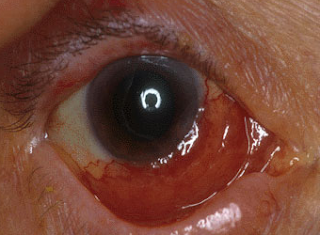
Lymphoma of the Ocular Adnexa
Some ocular lesions can be difficult to diagnose even with initial pathology reports. Primary Ocular Adnexal MALT Lymphoma (POAML) can sometimes produce a great deal of amyloid making Amyloidosis look like the diagnosis. A special test called flow cytometry will show monoclonal B cell aggregates if the diagnosis is MALT.
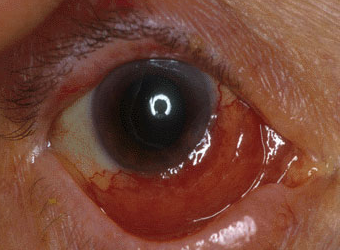
Depending on the stage of the POAML and a full body work up for the presence of lymphoma elsewhere, the prognosis is good in many cases.
The best doctors for POAML in the Washington DC area are:
Ocular Oncology
Akrit Sodhi, M.D. (Johns Hopkins Hospital and Columbia)
More information below.
Sandra Lora Cremers, MD, FACS
See full Wikipedia post at end of this page.
Blood. 2016 Oct 27. pii: blood-2016-05-714584. [Epub ahead of print]
Long term course of patients with primary ocular adnexal malt lymphoma: a large single institution cohort study.
Abstract
Extranodal Marginal Zone B-cell Lymphoma of the Ocular Adnexa.
Abstract
BACKGROUND:
METHODS:
RESULTS:
CONCLUSIONS:
Favourable response to rituximab by an ocular adnexal primary lymphoma.
Abstract
CASE REPORT:
DISCUSSION:
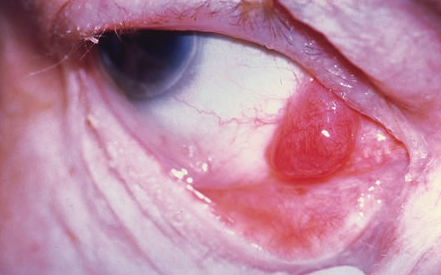
Salmon-coloured lesions mimicking conjunctival papillae: an unusual presentation of unilateral conjunctival lymphoma in a young man.
Author information
- 1Department of Optometry and Vision Sciences, The University of Melbourne, Victoria, Australia. ckwang@unimelb.edu.au.
- 2Harris, Blake and Parsons, Croydon, Victoria, Australia.
Introduction
Case Presentation



Discussion
Non-Hodgkin’s lymphoma subtypes involving the eye
Clinical features
Management
Conclusion
Financial support and sponsorship
Conflicts of interest
References
PUBLISHED 5 APRIL 2016
Better Options Emerging In Ocular Oncology
Current treatments are saving eyes and lives.
 |
| Figure 1. A) Small choroidal melanoma. B) Appearance one week after transvitreal fine-needle aspiration biopsy using a 25-ga. needle. |
Retinoblastoma
Representing 6.1 percent of all cancer in children younger than 5 years of age, retinoblastoma is the most common primary intraocular cancer in children. It primarily affects young children, with approximately 90 percent of cases occurring in children younger than 3 years of age. It is a highly malignant tumor that results in death in one to two years if left untreated. Worldwide, half of the children who are diagnosed with retinoblastoma present with extraocular manifestations that suggest advanced disease. This contributes to high mortality rates in developing countries (39 percent in Asia and 70 percent in Africa). Retinoblastoma confined to the globe, which is typical of cases diagnosed in the developed world, has survival rates as high as 96 percent to 100 percent. Treatment options include enucleation, external-beam radiation therapy, chemotherapy and focal therapies such as cryotherapy, laser therapy and brachytherapy.1
David Abramson, MD, says that in 90 percent of children with retinoblastoma, the disease is detected by a family member, usually their mother. They notice leukocoria or a whiteness or a glow in the eye, which is actually the cancer itself.
Two innovative treatments have caused a remarkable transformation in the treatment of retinoblastoma and have changed patients’ lives. “These are the use of intra-arterial chemotherapy and the use of intravitreal chemotherapy,” says Dr. Abramson, who is the chief of the ophthalmic oncology service at Memorial Sloan Kettering Cancer Center and a professor of ophthalmology at Weill-Cornell Medical School. “Historically, virtually all eyes in unilateral patients were enucleated as treatment. It is a good treatment in the sense that it removes the cancer, and more than 95 percent of the children survive, but it is a terrible treatment in the sense that you are removing a human eye, some of which have vision and others that may have the ability to regain vision after eye-salvaging therapy.”
Systemic chemotherapy has been used for retinoblastoma since 1953. “It is not new, and chemotherapy for intraocular disease has always caused a nice response, but it has never cured the tumors or prevented them from recurring,” says Dr. Abramson. “We’ve abandoned the use of systemic chemotherapy, which was never effective for the advanced eyes anyway. We’ve also abandoned radiation, which has many complications long term, especially second cancers; so there has truly been a transformation in management worldwide.
“In the past 10 years, since the introduction of intra-arterial chemotherapy, which Pierre Gobin, MD, and I did for the first time in May 2006, we’ve gone from a time when 95 percent of all children with unilateral retinoblastoma lost an eye, to a time when we are saving the eye in 95 percent of children with unilateral retinoblastoma. And, this has been accomplished without sacrificing life, which is important.”
Dr. Abramson and colleagues recently published a study demonstrating this.2 The study included 156 eyes of 156 retinoblastoma patients and showed that primary enucleation rates have progressively decreased from a rate of more than 95 percent before intra-arterial chemotherapy to 66.7 percent in the first year of intra-arterial chemotherapy treatment to the current rate of 7.4 percent. Additionally, the percentage of patients receiving intra-arterial chemotherapy has progressively increased from 33.3 percent in 2006 to 92.6 percent in 2014. And, there have been no cases of death from disease metastasis in those receiving intra-arterial chemotherapy, while two patients treated with primary enucleation died of metastatic disease.
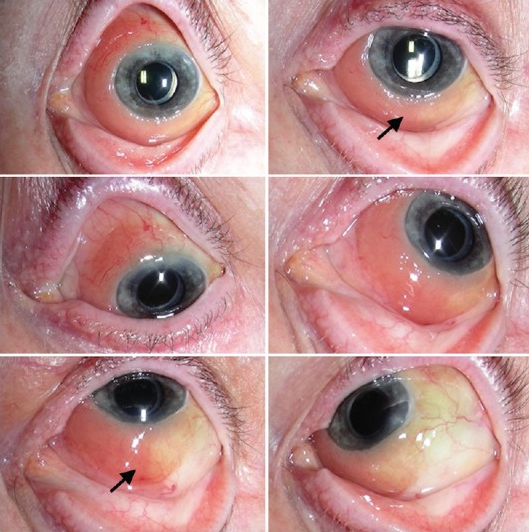
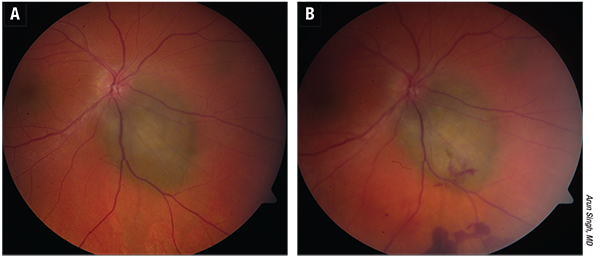
 |
| Figure 2. Ocular adnexal lymphoma (extranodal marginal zone type) A). Three months following treatment with 25 Gy external radiation. B) There is total regression of lymphoma without any surface secondary effects. (Images courtesy of Arun Singh, MD) |
Intra-arterial chemotherapy is administered by catheterizing a femoral artery, with the catheter passing from the abdominal aorta through the thoracic aorta through the internal carotid and then directly into the ophthalmic artery. Only a few ccs of chemotherapy are needed because the concentration in the eye is so high that it is very effective at destroying retinoblastoma. “An advantage is that the total dose is so low that the children don’t get sick or lose their hair,” says Dr. Abramson. “It is an outpatient procedure that takes an hour. The average child has it done either three or four times. Only about 1 percent of children develop fever or neutropenia or require blood transfusions. Half of the countries in the world that are using it are developing countries. That’s critical because, last year, half of the children in the world who developed retinoblastoma died. The main reason they died is that, in much of the world, when a family is told that the eye has to come out, they refuse and never go back to medical care. This is true throughout much of Asia and Africa, which is a large part of the world’s population. Children are not dying because the doctors don’t know how to treat it. It’s because the treatment, which does work, is unacceptable socially and culturally.”
Dr. Abramson and his colleagues recently conducted another study comparing enucleation with intra-arterial chemotherapy, and they found that there were more orbital recurrences of retinoblastoma in eyes primarily treated with enucleation. The study, which included 140 eyes in 135 patients with advanced retinoblastoma, found that treatment with intra-arterial chemotherapy did not increase the chance of orbital recurrence, metastatic disease or death compared with primary enucleation in these patients.3
The main reason that eyes have been removed for retinoblastoma is that retinoblastoma breaks off into little pieces called seeds, and the seeds have no blood supply and don’t divide very often, so they have always been resistant to chemotherapy and to radiation. “For eyes with advanced seeds, less than 25 percent could ever be saved with radiation,” Dr. Abramson says. “With systemic chemotherapy, almost none of them can be saved. Intra-arterial chemotherapy brought us from under 20 percent to about a 70 percent success rate. Then, Francis Munier, MD, in Switzerland introduced a modification of a technique that has also been around for 50 years. He introduced what we hope is a safe technique for injecting chemotherapy directly in the eye. It has increased the chance of saving eyes with vitreous seeds from near 0 percent to close to 90 percent. It does have some toxicity. You don’t get away without any complications.”
This new technique (intravitreous chemotherapy) and dosage achieved an unprecedented success rate of tumor control in the presence of vitreous seeding, and none of the treated eyes required external beam irradiation to control the vitreous seeding.4
This study included 23 eyes with active vitreous seeding that were eligible for intravitreous chemotherapy. These eyes received 122 intravitreal injections of melphalan (20 to 30 µg) given every seven to 10 days. All patients are alive and without evidence of extraocular spread. Eighty-seven percent of eyes achieved globe retention, and all retained eyes were in complete remission after a median follow-up period of 22 months. A localized peripheral salt-and-pepper retinopathy was observed at the injection site in 10 eyes.
Uveal Melanoma
Melanoma of the uveal tract is rare, but it is the most common primary intraocular malignancy in adults. It typically occurs in older adults, and a number of factors influence prognosis. These include cell type, tumor size, location of the anterior margin of the tumor, degree of ciliary body involvement and extraocular extension. The most commonly used predictor of outcome after enucleation is cell type, with spindle-A cell melanomas having the best prognosis and epithelioid cell melanomas having the worst prognosis.
“The majority of uveal melanomas are treated with a special type of radiation, delivered from a point source, known as brachytherapy. In contrast, if someone has a spread of cancer to their eye, such as from the lung or breast, it is generally treated with external beam radiation, as these tumors are more radiosensitive,” says William F. Mieler, MD, who is from the Department of Ophthalmology and Visual Sciences at the University of Illinois at Chicago.
According to Dr. Mieler, success rates of uveal melanoma treatment are quite good, as brachytherapy treatment offers approximately an 80 to 90 percent success rate in terms of tumor containment. “Uveal melanoma presentation ranges extensively from visual blurriness, to visual field defect, to no symptoms at all,” he says. “It depends on where the tumor is located inside the eye. Many tumors are found during a routine screening examination, especially if it is not involving the central region of the eye. When treating these patients, there are two main goals—keeping the patient alive, healthy and free of metastatic disease, and preserving vision, whenever possible.”
A recent study found that ruthenium-106 brachytherapy is useful for controlling uveal melanoma tumors, preserving the eye and preserving visual function.5 The study included 143 eyes, and the median follow-up was 37.9 months. Estimated local tumor recurrence rate was 3 percent at 12 months, 8.4 percent at 24 months and 14.7 percent at 48 months after irradiation. Likelihood of eye preservation was 97.7 percent, 94.7 percent and 91.8 percent at 12, 24 and 48 months respectively after irradiation.
The key to treating uveal melanoma is to prevent it from metastasizing, because after the development of metastatic disease, the rates of survival are the same for treated and untreated patients. “After prognostication, fine-needle aspiration and biopsy, patients with uveal melanoma are classified with regard to their risk for metastasis,” says Arun D. Singh, MD, director of the department of ophthalmic oncology at the Cole Eye Institute at the Cleveland Clinic. “The most significant change in how we practice and treat melanoma patients is incorporation of adjuvant therapy trials into our clinical practice.”
Currently, no adjuvant therapy regimen has shown promising results. Existing cytotoxic and immunotherapeutic regimens are being used in patients who have been identified as high-risk by tumor genetic criteria. Additionally, several novel cytotoxic, immune modulatory and targeted compounds that may potentially be used in the adjuvant setting are being studied in the metastatic setting. Preclinical studies have shown that methods that stimulate uveal melanoma cellular differentiation and/or dormancy are promising.6
“The diagnostic accuracy for melanomas is better than 99 percent, and while treatment is generally successful, work is ongoing to better identify patients who may be at higher risk for development of metastatic disease,” Dr. Mieler says. “Tissue analysis and genetic markers are allowing ophthalmologists to better assess patients regarding systemic risk. It’s not foolproof, but it is a major advance that is rapidly evolving.”
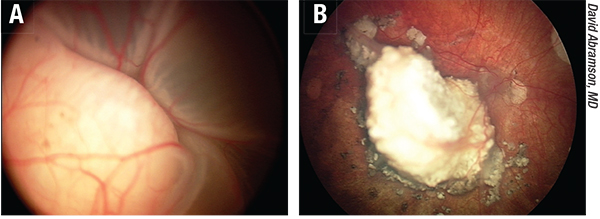 |
| Figure 3. A) Before and B) after successful intra-arterial chemotherapy treatment of retinoblastoma. The eye was blind at diagnosis and now has useful vision. |
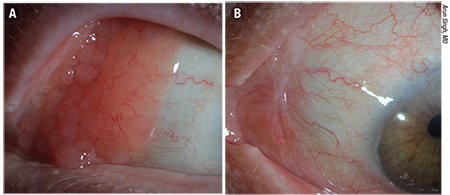
Ocular Adnexal Lymphoma
Another form of cancer is ocular adnexal lymphoma, which is a rare, primarily B-cell, non-Hodgkin’s lymphoma. Most are low-grade extranodal marginal zone (EMZL)/mucosa-associated lymphoid tissue (MALT) subtype. Before choosing a treatment approach, it is important to perform a thorough initial characterization of the disease extent and to follow staging procedures. Biopsy is the gold standard for diagnostic confirmation.7
Ocular adnexal lymphoma can involve the eyelids, conjunctiva, lacrimal apparatus, extraocular muscles and the orbit; however, most cases involve the orbit/lacrimal gland. A number of treatment options are available, and these include radiation, surgery, cryotherapy, chemotherapy, immunotherapy and antibiotic therapy. The standard primary therapy for ocular adnexal lymphoma is external-beam radiation therapy. EBRT has a high rate of success when used to treat ocular adnexal lymphoma without systemic involvement. Surgery is typically reserved for cases of isolated, highly focal conjunctival disease. Cryotherapy has also had limited success in eliminating disease, so it should be reserved for patients who cannot be treated any other way. With the exception of high-grade, aggressive, diffuse, large B-cell lymphoma, most cases of ocular adnexal lymphoma are localized; therefore, chemotherapy is not typically used. Recently, immunotherapy has been used as primary treatment, and rituximab, either alone or in combination with chemotherapy and/or radiation has shown promise for treating both bilateral ocular and systemic cases.7
“For lymphoma of the adnexa (marginal zone type), there are good data to indicate that radiation treatment is very effective and gives us very high control rates (more than 95 percent),” Dr. Singh adds. REVIEW
1. Chung CY, Medina CA, Aziz HA, Singh AD. Retinoblastoma: Evidence for stage-based chemotherapy. International Ophthalmology Clinics 2015;55(1):63-75.
2. Abramson DH, Fabius AW, Issa R, et al. Advanced unilateral retinoblastoma: The impact of ophthalmic artery chemosurgery on enucleation rate and patient survival at MSKCC. PLoS One 2015;10(12):e0145436.
3. Yannuzzi NA, Francis JH, Marr BP, et al. Enucleation vs ophthalmic artery chemosurgery for advanced intraocular retinoblastoma: A retrospective analysis. JAMA Ophthalmol 2015;133:1062-1066.
4. Munier FL, Gaillard MC, Balmer A, et al. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: From prohibition to conditional indications. Br J Ophthalmol doi:10.1136/bjophthalmol-2011-301450.
5. Tarmann L, Wackernagel W, Avian A, et al. Ruthenium-106 plaque brachytherapy for uveal melanoma. Br J Ophthalmol 2015;99(12):1644-1649.
6. Choudhary MM, Triozzi PL, Singh AD. Uveal melanoma: Evidence for adjuvant therapy. International Ophthalmology Clinics 2015;55(1):45-51.
7. Aronow ME. Ocular adnexal lymphoma: Evidence-based treatment approach. Int Ophthalmol Clin 2015 Winter:55(1):97-109.
MALT lymphoma
| MALT lymphoma | |
|---|---|

Endoscopic image of gastric MALT lymphoma taken in body of stomach in patient who presented with upper GI hemorrhage. Appearance is similar to gastric ulcerwith adherent clot.
|
|
| Classification and external resources | |
| Specialty | oncology |
| ICD–10 | C88.4 |
| ICD–9-CM | 200.3 |
| ICD-O | M9699/3 |
| OMIM | 604860 |
| DiseasesDB | 31339 |
| MeSH | D018442 |
Contents
[hide]
Diagnosis and staging[edit]
Associations[edit]
Treatment[edit]
Radiotherapy[edit]
Chemotherapy[edit]
- t(1;14)(p22;q32), which deregulates BCL10, at the locus 1p22.
- t(14;18)(q32;q21), which deregulates MALT1, at the locus 18q21.
Epidemiology[edit]
See also[edit]
References[edit]
- ^ Taal, B G; Boot, H; van Heerde, P; de Jong, D; Hart, A A; Burgers, J M (1 October 1996). “Primary non-Hodgkin lymphoma of the stomach: endoscopic pattern and prognosis in low versus high grade malignancy in relation to the MALT concept.”. Gut. 39 (4): 556–561. doi:10.1136/gut.39.4.556.
- ^ Janusz, edited by Jankowski, (2012). Handbook of Gastrointestinal Cancer (2 ed.). Chicester: John Wiley and Sons Ltd. pp. 243–244. ISBN 978-0-470-65624-2.
- ^ Wotherspoon, AC; Doglioni, C; Diss, TC; Pan, L; Moschini, A; de Boni, M; Isaacson, PG (4 September 1993). “Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori.”. Lancet. 342 (8871): 575–7. doi:10.1016/0140-6736(93)91409-f. PMID 8102719.
- ^ Parsonnet J, Hansen S, Rodriguez L, Gelb A, Warnke R, Jellum E, Orentreich N, Vogelman J, Friedman G (1994). “Helicobacter pylori infection and gastric lymphoma.”. N Engl J Med. 330 (18): 1267–71. doi:10.1056/NEJM199405053301803. PMID 8145781.
- ^ Hatakeyama, M.; Higashi, H. (2005). “Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis”. Cancer Science. 96: 835–843. doi:10.1111/j.1349-7006.2005.00130.x. PMID 16367902.
- ^ Bayerdorffer, E; Oertel, H; Lehn, N; Kasper, G; Mannes, G A; Sauerbruch, T; Stolte, M (1 August 1989). “Topographic association between active gastritis and Campylobacter pylori colonisation.”. Journal of Clinical Pathology. 42 (8): 834–839. doi:10.1136/jcp.42.8.834.
- ^ Park, Jeong Bae (2014). “infection in gastric mucosa-associated lymphoid tissue lymphoma”. World Journal of Gastroenterology. 20 (11): 2751. doi:10.3748/wjg.v20.i11.2751.
- ^ Fischbach, W; Goebeler, M E; Ruskone-Fourmestraux, A; Wundisch, T; Neubauer, A; Raderer, M; Savio, A (1 December 2007). “Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series”. Gut. 56 (12): 1685–1687. doi:10.1136/gut.2006.096420.
- ^ Sarah, Silverman. “MALT lymphoma Diagnosis, Staging, Treatment”. http://pylori.org/. UEG. External link in
|website=(help) - ^ Noels H, van Loo G, Hagens S, et al. (April 2007). “A Novel TRAF6 binding site in MALT1 defines distinct mechanisms of NF-kappaB activation by API2middle dotMALT1 fusions”. J. Biol. Chem. 282(14): 10180–9. doi:10.1074/jbc.M611038200. PMID 17287209.
- ^ Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, Ye H, Molina T, Bouhnik Y, Hamoudi R, Diss T, Dogan A, Megraud F, Rambaud J, Du M, Isaacson P (2001). “Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy.”. Lancet. 357 (9249): 39–40. doi:10.1016/S0140-6736(00)03571-6. PMID 11197361.
- ^ Tomita, N; Kodaira, T; Tachibana, H; Nakamura, T; Mizoguchi, N; Takada, A (February 2009). “Favorable outcomes of radiotherapy for early-stage mucosa-associated lymphoid tissue lymphoma.”. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 90 (2): 231–5. doi:10.1016/j.radonc.2008.12.004. PMID 19135751.
- ^ Schechter, NR; Portlock, CS; Yahalom, J (May 1998). “Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone.”. Journal of Clinical Oncology. 16 (5): 1916–21. PMID 9586910.
- ^ Avilés, A; Nambo, MJ; Neri, N; Talavera, A; Cleto, S (2005). “Mucosa-associated lymphoid tissue (MALT) lymphoma of the stomach: results of a controlled clinical trial.”. Medical oncology (Northwood, London, England). 22 (1): 57–62. doi:10.1385/mo:22:1:057. PMID 15750197.
- ^ Conconi, A; Martinelli, G; Thiéblemont, C; Ferreri, AJ; Devizzi, L; Peccatori, F; Ponzoni, M; Pedrinis, E; Dell’Oro, S; Pruneri, G; Filipazzi, V; Dietrich, PY; Gianni, AM; Coiffier, B; Cavalli, F; Zucca, E (15 October 2003). “Clinical activity of rituximab in extranodal marginal zone B-cell lymphoma of MALT type.”. Blood. 102 (8): 2741–5. doi:10.1182/blood-2002-11-3496. PMID 12842999.
- ^ Martinelli, G; Laszlo, D; Ferreri, AJ; Pruneri, G; Ponzoni, M; Conconi, A; Crosta, C; Pedrinis, E; Bertoni, F; Calabrese, L; Zucca, E (20 March 2005). “Clinical activity of rituximab in gastric marginal zone non-Hodgkin’s lymphoma resistant to or not eligible for anti-Helicobacter pylori therapy.”. Journal of Clinical Oncology. 23 (9): 1979–83. doi:10.1200/jco.2005.08.128. PMID 15668468.
- ^ Lévy, M; Copie-Bergman, C; Gameiro, C; Chaumette, MT; Delfau-Larue, MH; Haioun, C; Charachon, A; Hemery, F; Gaulard, P; Leroy, K; Delchier, JC (1 August 2005). “Prognostic value of translocation t(11;18) in tumoral response of low-grade gastric lymphoma of mucosa-associated lymphoid tissue type to oral chemotherapy.”. Journal of Clinical Oncology. 23 (22): 5061–6. doi:10.1200/jco.2005.05.660. PMID 16051953.
- ^ Zinzani, PL; Stefoni, V; Musuraca, G; Tani, M; Alinari, L; Gabriele, A; Marchi, E; Pileri, S; Baccarani, M (15 May 2004). “Fludarabine-containing chemotherapy as frontline treatment of nongastrointestinal mucosa-associated lymphoid tissue lymphoma.”. Cancer. 100 (10): 2190–4. doi:10.1002/cncr.20237. PMID 15139063.
- ^ Jager, G; Neumeister, P; Quehenberger, F; Wohrer, S; Linkesch, W; Raderer, M (6 April 2006). “Prolonged clinical remission in patients with extranodal marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type treated with cladribine: 6 year follow-up of a phase II trial”. Annals of Oncology. 17 (11): 1722–1723. doi:10.1093/annonc/mdl126.
- ^ Zucca, E; Conconi, A; Laszlo, D; López-Guillermo, A; Bouabdallah, R; Coiffier, B; Sebban, C; Jardin, F; Vitolo, U; Morschhauser, F; Pileri, SA; Copie-Bergman, C; Campo, E; Jack, A; Floriani, I; Johnson, P; Martelli, M; Cavalli, F; Martinelli, G; Thieblemont, C (10 February 2013). “Addition of rituximab to chlorambucil produces superior event-free survival in the treatment of patients with extranodal marginal-zone B-cell lymphoma: 5-year analysis of the IELSG-19 Randomized Study.”. Journal of Clinical Oncology. 31 (5): 565–72. doi:10.1200/jco.2011.40.6272. PMID 23295789.
- ^ Ye H, Gong L, Liu H, et al. (February 2005). “MALT lymphoma with t(14;18)(q32;q21)/IGH-MALT1 is characterized by strong cytoplasmic MALT1 and BCL10 expression”. J. Pathol. 205 (3): 293–301. doi:10.1002/path.1715. PMID 15682443.
- ^ Turgeon, Mary Louise (2005). Clinical hematology: theory and procedures. Hagerstown, MD: Lippincott Williams & Wilkins. p. 283. ISBN 0-7817-5007-5.
Frequency of lymphoid neoplasms. (Source: Modified from WHO Blue Book on Tumour of Hematopoietic and Lymphoid Tissues. 2001, p. 2001.)